
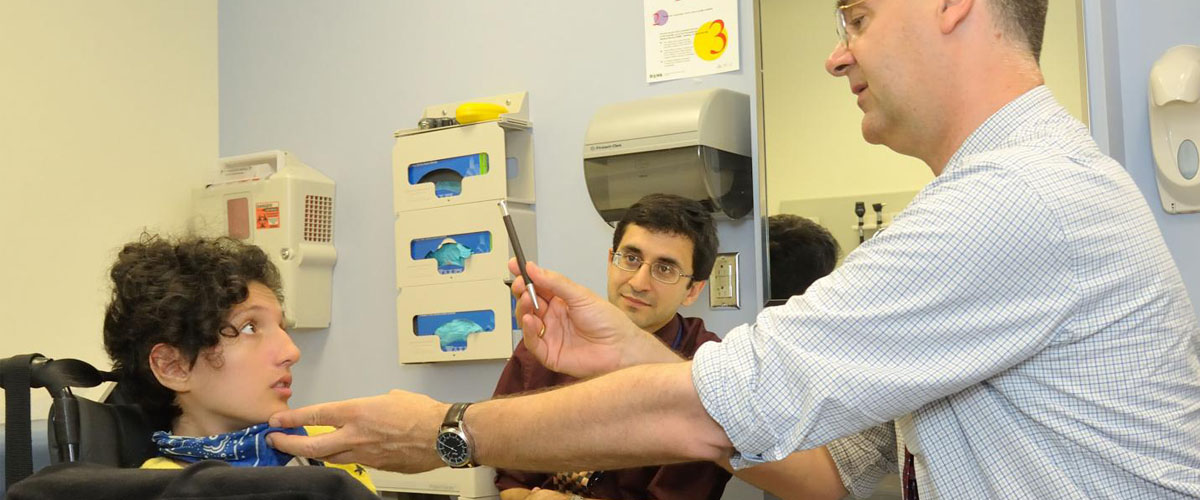
Amyotrophic Lateral Sclerosis (ALS) is a rare neurodegenerative syndrome, which is characterized by the progressive loss of the motor neurons in the brain and spinal cord that leads to grave impairments involuntary muscle movement. It is believed to be a disease of older adults, is now been identified in children as young as 3 years old. To answer the question, as to why it appeared in younger people, researchers have now identified some mutations in the genes that cause the early onset.
ALS affects the muscles by decreasing muscle power and muscle mass throughout the body, impairing activities of daily living like walking, talking, and eventually breathing, thus causing death in most of its patients within a year of manifesting symptoms.
This disease has an unknown origin due to its sporadic nature in people aged between 55 to 75. It has a different manifestation in different people, depending upon the genetics, it can, however, rarely show up in younger people as identified by a group of scientists from the USA. They found that these rare displays of disease concurring with the uncommon set of mutations associated with a distinct form of genetic ALS in children, which has been a puzzle for doctors for years.
An international investigation of 10 young patients manifesting ALS-like symptoms in early childhood, lead to the identification of the genetic basis of this rare disorder. The point of interest for taking such children is the early onset of disease and slower progression of symptoms, even though these young patients manifest problems in an upper and lower motor neuron which is suggestive of ALS, explained Payam Mohassel from the National Institute of Health.
By studying the DNA of 10 young patients in the study they found that the reason for the early onset of ALS in the younger population is the set of genetic mutation in the gene SPTLC1, that encodes for a protein, which in turn works as a catalyst for the production of sphingolipids – the fatty molecules.
Interestingly four patients in the study, who are from one family seems to have inherited their genetic variations of the SPTLC1 gene from one of their parents, whereas, the other six patients in the study, that weren’t related to one another, seems to have random de novo mutation in the gene, means it appeared for the first time in the family. In all the cases, the mutation is what leads to the excessive production of sphingolipids, linked to abnormally elevated levels of an enzyme that helps make the lipids, known as serine palmitoyltransferase or SPT. These mutations simply short-circuit the feedback loop that controls SPT. The oversupply of sphingolipids accumulates in motor neurons, causing the early onset of ALS.
In the past, abnormality in the SPTLC1 gene is linked to other neurodegenerative disorders like Hereditary Sensory and Autonomic Neuropathy. When the scientists could turn off this mutation by using small interfering RNA (siRNA) to target the mutant allele, they could potentially prevent this all from happening as it will inhibit overactive SPT production.
This study is a breakthrough for the treatment of early-onset ALS in children as now the precision gene silencing method could be used as a treatment method.









Add comment